Published 12 November 2025 in Journal of Structural Biology X (doi 10.1016/j.yjsbx.2025.100139):
Calcium stabilizes the flexible N-terminal domain of the bacterial ion channel DeCLIC
Chen Fan,° Marie Lycksell, Yuxuan Zhuang, Rebecca J Howard,° Erik Lindahl°
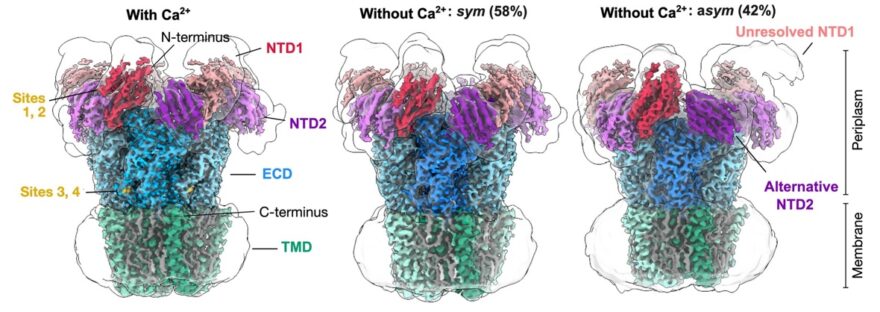
Pentameric ligand-gated ion channels (pLGICs) are responsible for the rapid conversion of chemical to electrical signals. In addition to the canonical extracellular and transmembrane domains, some prokaryotic pLGICs contain an N-terminal domain (NTD) of unclear structure and function. In one such case, the calcium-sensitive channel DeCLIC, the NTD appears to accelerate gating; however, its evident flexibility has posed a challenge to model building, and its role in calcium sensitivity is unclear. Here we report cryo-EM structures of DeCLIC in circularized lipid nanodiscs, achieving the highest resolution reported so far, and enabling definition of calcium-binding sites in both the N-terminal and canonical extracellular domains. In addition to the symmetric state, calcium depletion promoted an asymmetric conformation of the NTD, offering a structural rationale for small-angle scattering results. Behavior of these structures in molecular dynamics simulations demonstrated calcium stabilization of the NTD. These features of DeCLIC offer a model system for ion-channel modulation by a flexible accessory domain, potentially conserved in structurally homologous systems across evolution.
°Co-corresponding authors
Read the full publication here.