Published 2 September 2020 in Nature (doi 10.1038/s41586-020-2654-5):
Shared structural mechanisms of general anaesthetics and benzodiazepines
Jeong Joo Kim, Anant Gharpure, Jinfeng Teng, Yuxuan Zhuang, Rebecca J Howard, Shaotong Zhu, Colleen M Noviello, Richard M Walsh Jr, Erik Lindahl, Ryan E Hibbs
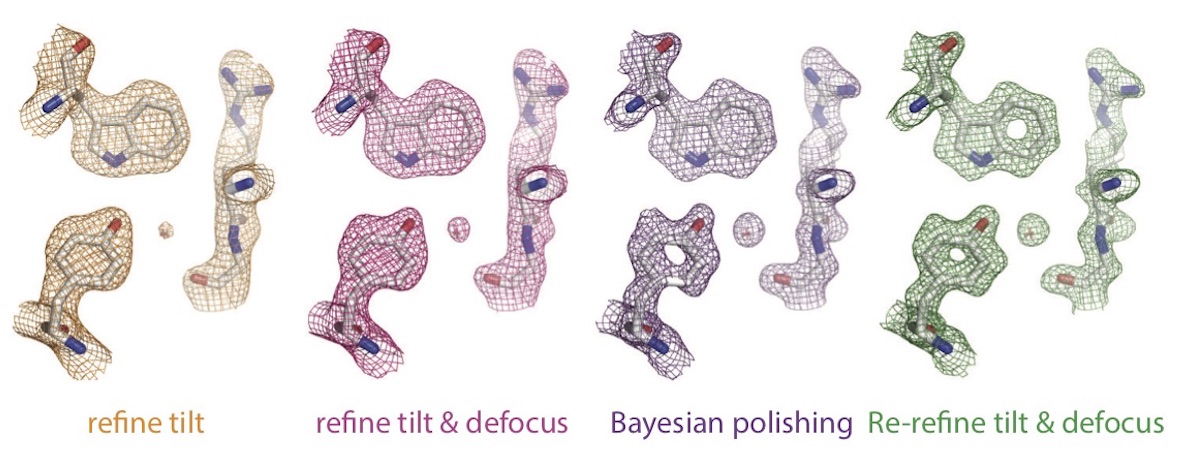
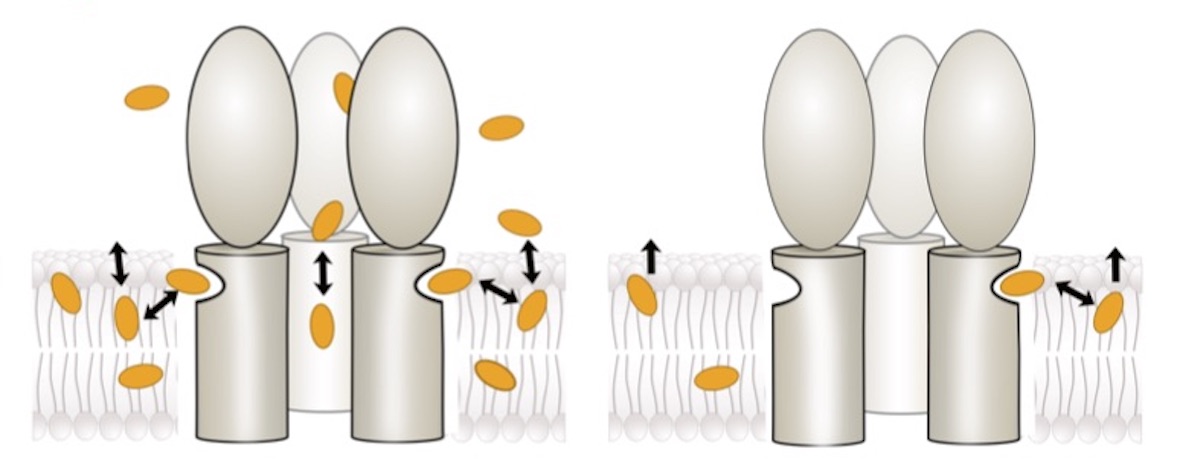
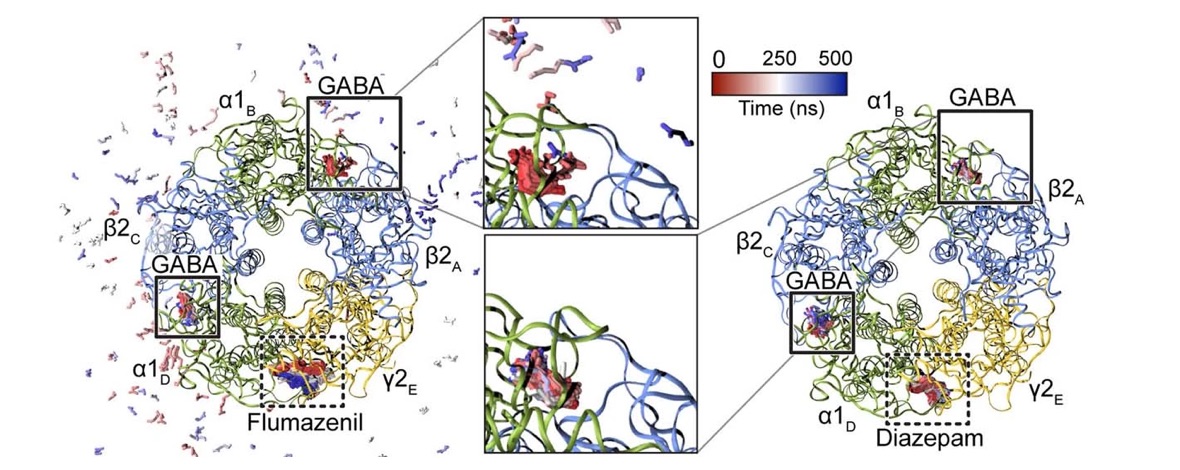
Most general anaesthetics and classical benzodiazepine drugs act through positive modulation of γ-aminobutyric acid type A (GABAA) receptors to dampen neuronal activity in the brain. However, direct structural information on the mechanisms of general anaesthetics at their physiological receptor sites is lacking. Here we present cryo-electron microscopy structures of GABAA receptors bound to intravenous anaesthetics, benzodiazepines and inhibitory modulators. These structures were solved in a lipidic environment and are complemented by electrophysiology and molecular dynamics simulations. Structures of GABAA receptors in complex with the anaesthetics phenobarbital, etomidate and propofol reveal both distinct and common transmembrane binding sites, which are shared in part by the benzodiazepine drug diazepam. Structures in which GABAA receptors are bound by benzodiazepine-site ligands identify an additional membrane binding site for diazepam and suggest an allosteric mechanism for anaesthetic reversal by flumazenil. This study provides a foundation for understanding how pharmacologically diverse and clinically essential drugs act through overlapping and distinct mechanisms to potentiate inhibitory signalling in the brain.
Read the full publication here.