Available online 19 June 2021 in the Journal of Biological Chemistry (doi 10.1016/j.jbc.2021.100899):
Regulation of a pentameric ligand-gated ion channel by a semi-conserved cationic-lipid binding site
Akshay Sridhar*, Sarah CR Lummis*, Diletta Pasini, Aujan Mehregan, Marijke Brams, Kumiko Kambara, Daniel Bertrand, Erik Lindahl, Rebecca J Howard, Chris Ulens
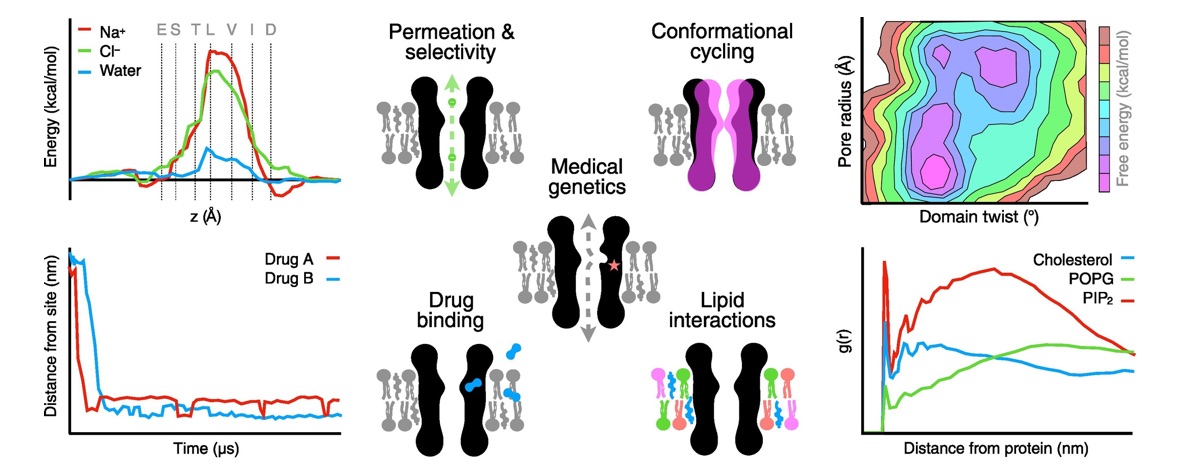
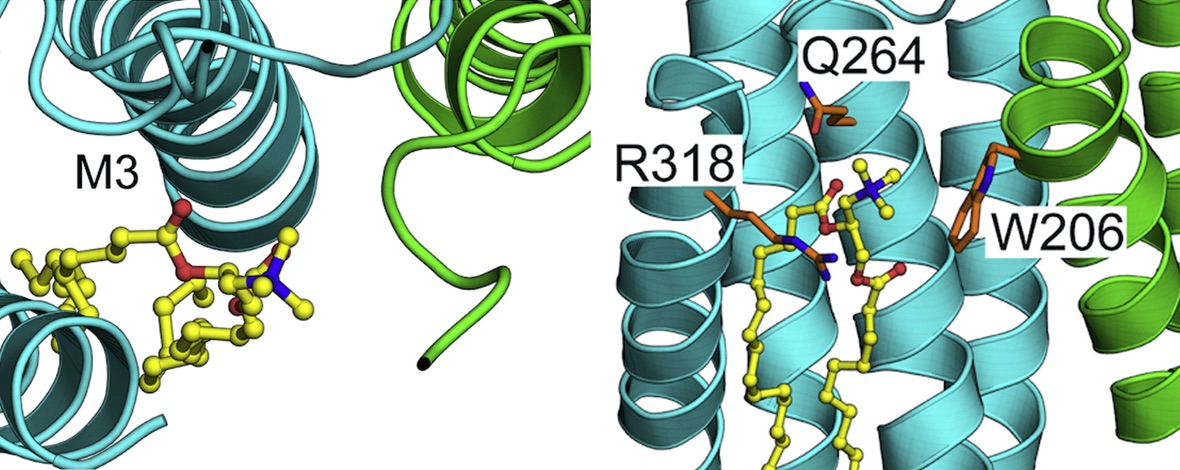
Pentameric ligand-gated ion channels (pLGICs) are crucial mediators of electrochemical signal transduction in various organisms from bacteria to humans. Lipids play an important role in regulating pLGIC function, yet the structural bases for specific pLGIC-lipid interactions remain poorly understood. The bacterial channel ELIC recapitulates several properties of eukaryotic pLGICs, including activation by the neurotransmitter GABA, and binding and modulation by lipids, offering a simplified model system for structure-function relationship studies. In this study, functional effects of non-canonical amino acid substitution of a potential lipid-interacting residue (W206) at the top of the M1-helix, combined with detergent interactions observed in recent X-ray structures, are consistent with this region being the location of a lipid binding site on the outward face of the ELIC transmembrane domain. Coarse-grained and atomistic molecular dynamics simulations revealed preferential binding of lipids containing a positive charge, particularly involving interactions with residue W206, consistent with cation-π binding. Polar contacts from other regions of the protein, particularly M3 residue Q264, further support lipid binding via headgroup ester linkages. Aromatic residues were identified at analogous sites in a handful of eukaryotic family members, including the human GABAA receptor ε subunit, suggesting conservation of relevant interactions in other evolutionary branches. Further mutagenesis experiments indicated that mutations at this site in ε-containing GABAA receptors can change the apparent affinity of the agonist response to GABA, suggesting a potential role of this site in channel gating. In conclusion, this work details type-specific lipid interactions, which adds to our growing understanding of how lipids modulate pLGICs.
Read the full publication here.
*equal contributions